Peax
Project details
Released 9/15/2018
Peax is a tool for interactive concept learning and exploration of epigenomic patterns based on unsupervised machine learning with autoencoders.
GitHub repoA pattern explorer for epigenomic data.
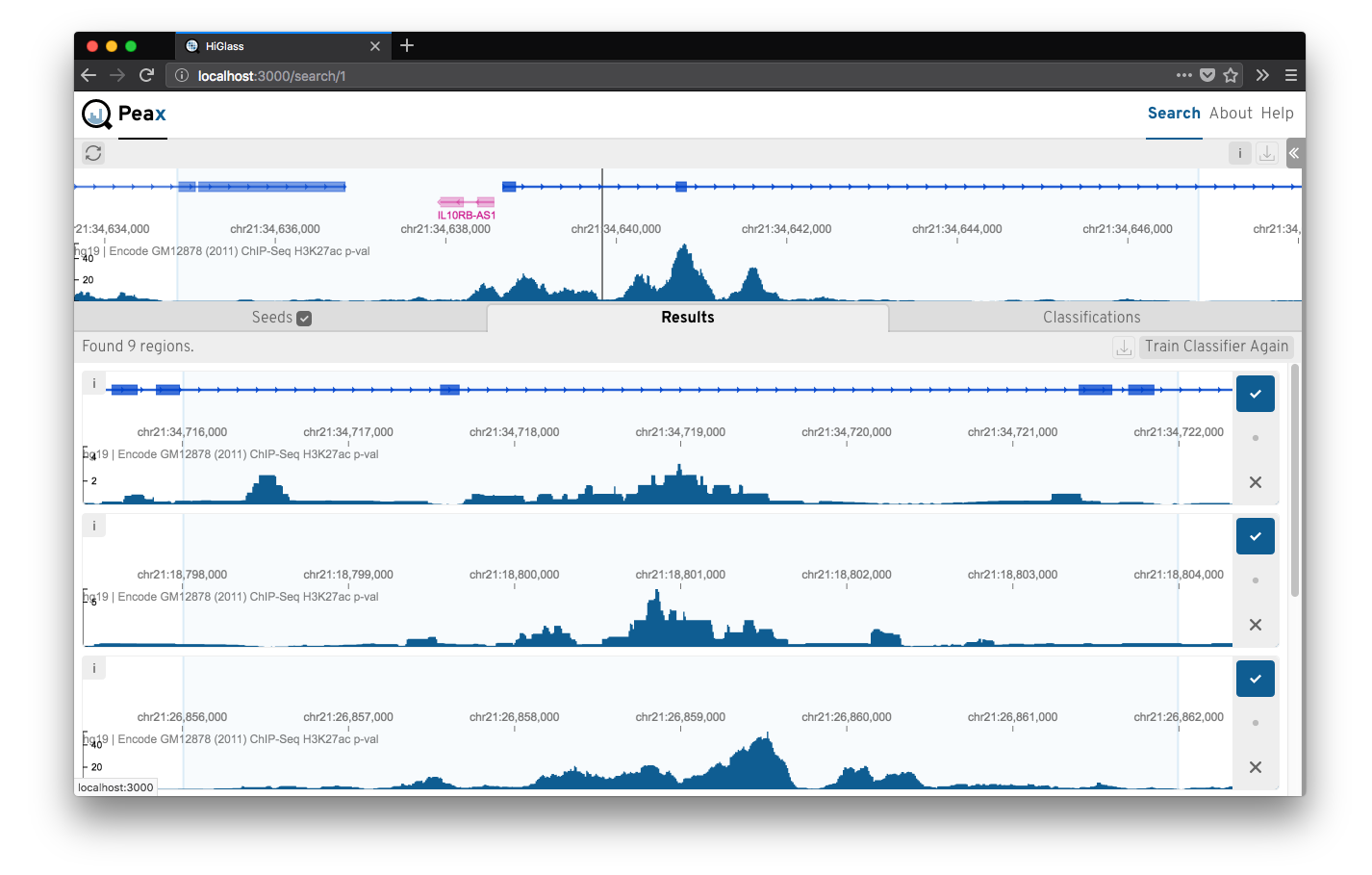
Epigenomic data expresses a rich body of diverse patterns that help to identify regulatory elements like promoter, enhancers, etc. But finding these patterns reliably genome wide is challenging. Peax is a tool for interactive visual pattern search and exploration of epigenomic patterns based on unsupervised representation learning with convolutional autoencoders. The visual search is driven by manually labeled genomic regions for actively learning a classifier to reflect your notion of interestingness. More at peax.lekschas.de.
Installation
git clone https://github.com/Novartis/peax peax && cd peax
make installDo not fear, make install is just a convenience function for setting up conda and installing npm packages.
Notes:
- If you're a macOS user you might need to brew install
libpngandopensslfor the pybbi package (see here) andxzfor pysam (if you see an error related tolzma.h).
Overview
Peax consists of three main parts:
- A server application for serving genomic and autoencoded data on the web. [/server].
- A user interface for exploring, visualizing, and interactively labeling genomic regions. [/ui].
- A set of examples showing how to configure Peax and build your own. [/examples]
Data
We provide 6 autoencoders trained on 3 kb, 12 kb, and 120 kb window sizes (with 25, 100, and 1000 bp binning) on DNase-seq and histone mark ChIP-seq data.
You can find the autoencoder at zenodo.org/record/2609763.
Preprint
Lekschas et al., Peax: Interactive Visual Pattern Search in Sequential Data Using Unsupervised Deep Representation Learning, bioRxiv, 2019, doi: 10.1101/597518.
Quick start
Peax comes with 6 autoencoders for DNase-seq and histone mark ChIP-seq data and several example configurations for which we provide convenience scripts to get you started as quickly as possible.
For instance, run one of the following commands to start Peax with a DNase-seq track for 3 kb, 12 kb, and 120 kb genomic windows.
| Command | Window Size | Step Freq. | Chromosomes |
|---|---|---|---|
make example-3kb | 3 kb | 2 | 21 |
make example-12kb | 12 kb | 3 | 20-21 |
make example-120kb | 120 kb | 6 | 17-21 |
Note: The first time Peax is started it will precompute the datasets for exploration. This can take a few minutes depending on your hardware. Also, these demos will only prepare the above mentioned chromosomes, so don't try to search for patterns on another chromosome. It won't work! For your own data you can freely configure this of course.
The scripts will download test ENCODE tracks and use the matching configuration to start the server. More examples are described in /examples.
Get Started
In the following we describe how you can configure Peax for your own data.
Configure Peax with your data
Next, you need to configure Peax with your data to tell it which tracks you want to visualize in HiGlass and which of those tracks are encodable using an (auto)encoder.
The fastest way to get started is to copy the example config:
cp config.json.sample config.jsonThe config file has 6 top level properties:
| Field | Description | Dtype |
|---|---|---|
| encoders | List of encoders. | list |
| datasets | List of tracks. | list |
| coords | Genome coordinates. Peax currently supports hg19, hg28, mm9, and mm10 | str |
| chroms | Chromosomes to to be searched. If omitted all chromosomes will be prepared for searching. | list |
| step_freq | Step frequency of the sliding window approach. E.g., given an encoder with window size 12 kb, a step frequency of 6 means that every 2 kb a 12 kb window will be extracted from the bigWig. | int |
| db_path | Relative path to the sqlite db for storing searches. | str |
The main parts to adjust are encoders and datasets. encoders is a list of (auto)encoder definitions for different datatypes.T here are two ways to configure an (auto)encoder: (a) point to a pre-defined autoencoder or (b) configure from scratch.
Assuming you want to use a predefined autoencoder all you have to do is
| Field | Description | Dtype |
|---|---|---|
| content_type | Unique string identifying the autoencoder in the configuration file | str |
| from_file | Relative path to the encoder configuration file. | str |
Example:
{
"content_type": "histone-mark-chip-seq-3kb",
"from_file": "examples/encoders.json"
}The encoder configuration file is a dictionary with the top level keys acting as the identifier and need to match content_type above. Given the example from above the file could look like this:
{
"histone-mark-chip-seq-3kb": {},
"dnase-seq-3kb": {}
}See [encoders.json](encoders.json) for an example. The specific definition if an autoencoder is the same as described in the following.
To configure an autoencoder from scratch you need to provide a dictionary with the following required format:
| Field | Description | Defaults | Dtype |
|---|---|---|---|
| autoencoder | Relative path to your pickled autoencoder model. (hdf5 file) | str | |
| encoder | Relative path to your pickled encoder model. (hdf5 file) | str | |
| decoder | Relative path to your pickled decoder model. (hdf5 file) | str | |
| content_type | Unique string describing the content this autoencoder can handle. Data tracks with the same content type will be encoded by this autoencoder. | str | |
| window_size | Window size in base pairs used for training the autoencoder. | int | |
| resolution | Resolution or bin size of the window in base pairs. | int | |
| latent_dim | Number of latent dimensions of the encoded windows. | int | |
| input_dim | Number of input dimensions for Keras. For 1D data these are 3: samples, data length (which is window_size / resolution), channels. | 3 | int |
| channels | Number of channels of the input data. This is normally 1. | 1 | int |
Note that if you have specified an autoencoder you do not need to provide separate encoder and decoder models.
Example:
{
"encoder": "path/to/my-12kb-chip-seq-encoder.h5",
"decoder": "path/to/my-12kb-chip-seq-decoder.h5",
"content_type": "histone-mark-chip-seq",
"window_size": 12000,
"resolution": 100,
"channels": 1,
"input_dim": 3,
"latent_dim": 12
}Datasets require the following format:
| Field | Description | Dtype |
|---|---|---|
| filepath | Relative path to the data file (bigWig or bigBed). | str |
| content_type | Unique string describing the content this dataset. If you want to search for patterns in this track you need to have an autoencoder with a matching content type. | str |
| id | A unique string identifying your track. (Optional) | str |
| name | A human readable name to be shown in HiGlass. (Optional) | str |
Example:
{
"filepath": "data/chip-seq/my-fancy-gm12878-chip-seq-h3k27ac-fc-signal.bigWig",
"content_type": "histone-mark-chip-seq",
"uuid": "my-fancy-gm12878-chip-seq-h3k27c-track",
"name": "My Fancy GM12878 ChIP-Seq H3k27c Track"
}Start Peax
First, start the Peax server to serve your data.
Note: The first time you run Peax on a new dataset all the data will be prepared! Depending on your machine this can take some time. If you want to track the progress activate the debugging mode using -d.
python start.pyNow go to http://localhost:5000.
To start.py script supports the following options:
usage: start.py [-h] [-c CONFIG] [--clear] [--clear-cache]
[--clear-cache-at-exit] [--clear-db] [-d] [--host HOST]
[--port PORT] [-v]
Peak Explorer CLI
optional arguments:
-h, --help show this help message and exit
-c CONFIG, --config CONFIG
path to your JSON config file
--clear clears the cache and database on startup
--clear-cache clears the cache on startup
--clear-cache-at-exit
clear the cache on shutdown
--clear-db clears the database on startup
-d, --debug turn on debug mode
--host HOST customize the hostname
--port PORT customize the port
-v, --verbose turn verbose logging onThe hostname defaults to localhost and the port of the backend server defaults to 5000.
In order to speed up subsequend user interaction, Peax initially prepapres all the data and caches that data under /cache. You can always remove this directory manually or clear the cache on startup or at exist using the --clear as specified above.
Development
Handy commands to keep in mind:
make installinstalls the conda environment and npm packages and builds the UImake updateupdates the conda environment and npm packages and rebuilds the UImake buildbuilds the UI./start.pystarts the Flask server application for serving data- [/ui]:
npm installinstalls and updates all the needed packages for the frontend - [/ui]:
npm buildcreates the production built of the frontend - [/ui]:
npm startstarts a dev server with hot reloading for the frontend
To start developing on the server and the ui in parallel, first start the backend server application using ./start.py and then start the frontend server application from ./ui using npm start. Both server's watch the source code, so whenever you make changes to the source code the servers will reload.
Configuration
There are 2 types of configuration files. The backend server configuration defines the datasets to explore and is described in detail above.
Additionally, the frontend application can be configured to talk to a different backend server and port if needed. Get started by copying the example configuration:
cd ui && cp config.json.sample config.jsonBy default the server is dynamically set to the hostname of the server running the frontend application. I.e., it is assumed that the backend server application is running on the same host as the frontend application. The port of the server defaults to 5000.
Start the backend and frontend apps
For development the backend and frontend applications run as seperate server applications.
# Backend server
./start.py --debug --config path/to/your/config.json
# Frontend server
cd ui && npm startInterested in NIBR Engineering?
At NIBR, you'll be at the forefront of technology — helping to shape it, develop it, and make it impactful. Partnering with scientists, our engineers create cutting-edge, state-of-the-art solutions that accelerate drug discovery and ultimately improve patients’ lives.
Learn more