Yet Another Pipeline
Project details
Released 11/6/2014
YAP is an extensible parallel framework, written in Python using OpenMPI libraries. It allows researchers to quickly build high throughput big data pipelines without extensive knowledge of parallel programming. The user interacts with the framework through simple configuration files to capture analysis parameters and user directed metadata, enabling reproducible research. Using YAP, analysts have been able to achieve a significant speed up of up to 36× in RNASeq workflow execution time.
YAP has been designed to be scalable and flexible. We have implemented YAP with a focus on next-generation sequencing (NGS), to meet the large data processing challenges at NIBR. However, the framework can be easily adapted for any kind of analysis. It can be executed on your local Linux workstations or large HPC cluster systems. The framework achieves efficiency by implementing optimal data handling mechanisms such as, parallel data distribution, avoiding file I/O using data streams and named pipes.
GitHub repoYAP compared to analysts' scripts
| Analysis | Data size | Number of cores | Analyst methods (hrs) | YAP (hrs) | Speed-up |
|---|---|---|---|---|---|
| RNASeq QC and Counts | 3 billion reads (150 samples) | 500 | 325.6 | 9 | 36× |
| Bacterial studies using Mothur | 230,000 reads | 72 | 90 | 12 | 8× |
| ChIPSeq Peak Calls | 190 million reads (6 samples) | 12 | 9.3 | 4.5 | 2× |
| EQP | 400 million reads (5 samples) | 60 | 45 | 12 | 4× |
| Traditional method | YAP | |
|---|---|---|
| I/O steps | 1400 file reads 1200 file writes | 200 file reads 800 file writes |
| Jobs spawned | 1500 | 1 MPI job |
| File-based reads reduced by 70% File-based writes reduced by 30% | ||
Example Analysis Output
The following images are the results of various applications run within the YAP framework, such as FastQC, FastQScreen, PicardTools, etc.
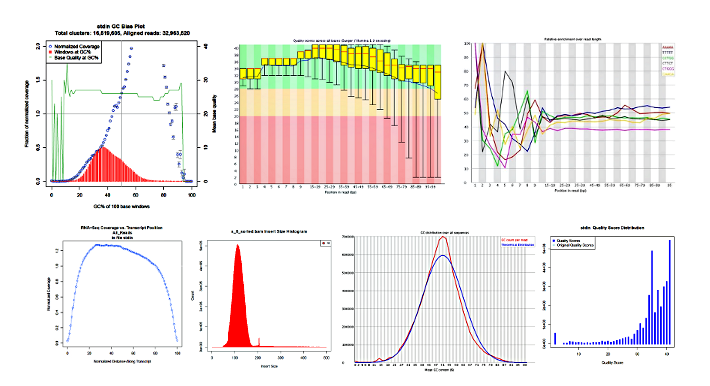
YAP consolidates results from across the samples for the various packages, such as gene counts from HTSeq and normalized counts from Cufflinks.
HTSeq gene counts
| SAMPLE | CR560274_1 | CR560457_1 | CR560502_1 | CR560562_1 | ... |
|---|---|---|---|---|---|
| NM_000014 | 34 | 13 | 35 | 34 | |
| NM_000015 | 2 | 1 | 1 | 1 | |
| NM_000016 | 0 | 0 | 0 | 0 | |
| NM_000017 | 27 | 11 | 9 | 3 | |
| NM_000018 | 0 | 0 | 0 | 0 | |
| NM_000019 | 18 | 48 | 17 | 14 | |
| NM_000020 | 0 | 0 | 0 | 0 | |
| NM_000021 | 0 | 0 | 0 | 0 | |
| ⋮ | ⋱ |
Normalized counts from Cufflinks
| SAMPLE | CR560274_1 | CR560457_1 | CR560502_1 | ... | |||
| TRACKING_ID | FPKM | FPKM_Status | FPKM | FPKM_Status | FPKM | FPKM_Status | |
| NM_000014 |chr12:9220303-9268558| | 0.187039 | OK | 0 | OK | 0.134608 | OK | |
| NM_000015 |chr8:18248754-18258723| | 0.218917 | OK | 0.152739 | OK | 0.13776 | OK | |
| NM_000016 |chr1:76190042-76229355| | 2.02618 | OK | 8.25528 | OK | 1.7346 | OK | |
| NM_000017 |chr12:121163570-121177811| | 1.40608 | OK | 0.980779 | OK | 0.708088 | OK | |
| ⋮ | ⋱ | ||||||
Reproducible research
Here's an example of the metadata automatically collected during a YAP run. By storing the commands and parameters used to run the job, YAP allows scientists to reproduce their analysis results at later points.
------------------------------ YAP ANALYSIS SUMMARY FOR WORKFLOW = yap2.3_test ------------------------------
Operating System Information= Linux yourhostname 2.6.18-371.9.1.el5#1 SMP Tue May 13 06:52:49 EDT 2014x86_64
USER= my_user
YAP SOURCE= /YAP/opensource/
Python Source= 2.7.5 (default, Sep 10 2013, 17:21:36) [GCC 4.1.2 20080704 (Red Hat 4.1.2-54)]
Analysis Start Time For Workflow : yap2.3_test 2014/09/04 15:55:44
YAP analysis general metadata:
1.comment:120 chars
2.analyst_name:120 chars
3.organisation_name:NIBR
Instrument Type= Illumina
Specimen Information= [tissue type]
Workflow type= rnaseq
Number of input files= 2
Number of processors= 6
Input files path for the workflow= /examples/sample_input
Input file provided:
1.RN0000108D_1 => /examples/sample_input/RN0000108D_1.fq
/examples/sample_input/RN0000108D_2.fq
Output file path for the workflow= /test_output/yap2.3_test
Sequence data type= paired end
Input file format= fastq
Maximum read length= 150
File chunk size (in megabytes)= 1024
Data distribution method=chunk_based
Output file path= /test_output/
-------------------------------------------------------------------------------------------------------------
Analysis stages :
Preprocess analysis= yes
Reference Sequence Alignment=yes
Postprocess Analysis= yes
-------------------------------------------------------------------------------------------------------------
Preprocess Analysis commands:
Barcodes information:
no_barcode_specified :
1. command name= fastq_screen,command line= /packages/FastQScreen/v0.4.1/fastq_screen --subset 500000 --paired --outdir output_directory --conf fastq_screen_v0.4.1.conf --aligner bowtie
2. command name= fastqc,command line= /packages/fastqc/0.10.1/fastqc --outdir output_directory --extract --threads 12
-------------------------------------------------------------------------------------------------------------
Aligner commands:
1. command name= bowtie,command line= /packages/bowtie/1.0.0/bowtie /accessory_files/indexes/bowtie/hg19 -q -v 2 -k 10 -m 10 --best -S -p 8 -1 pipe1 -2 pipe2 >output_file.sam
Alignment output data sort order= both
-------------------------------------------------------------------------------------------------------------
Samples re-grouped in this workflow:
None.
-------------------------------------------------------------------------------------------------------------
Potprocess analysis commands:
1. command type= :begin
command input : ['input_file_type *junctions.bed*', 'input_directory aligner_output']
1. command name= yap_junction_count,command line= yap_junction_count -exon_coordinates_file /accessory_files/human-ucsc-final_exon_coord.bed -exon_CoordToNumber_file /accessory_files/human-ucsc-final_exon_coord_number.bed -i - -o output_file
2. command type= :begin
command input : ['input_file_type *queryname*.sam', 'input_directory aligner_output']
1. command name= htseq-count,command line= /packages/python/2.6.5_gnu/bin/htseq-count -s no -q file_based_input /accessory_files/human-ucsc-refGene.gtf >output_file.out
3. command type= :begin_tee
command input : ['input_directory aligner_output', 'input_file_type *coordinate*']
1. command name= yap_exon_count,command line= yap_exon_count -f 1.0 -exon_coordinates_file /accessory_files/human-ucsc-final_exon_coord.bed -exon_CoordToNumber_file /accessory_files/human-ucsc-final_exon_coord_number.bed -i - -o output_file
2. command name= CollectAlignmentSummaryMetrics,command line= java -Xmx1g -jar /packages/picard-tools/1.89/CollectAlignmentSummaryMetrics.jar VALIDATION_STRINGENCY= SILENT I= /dev/stdin O= output_file.txt IS_BISULFITE_SEQUENCED= true ASSUME_SORTED= True REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa QUIET= True
3. command name= QualityScoreDistribution,command line= java -Xmx1g -jar /packages/picard-tools/1.89/QualityScoreDistribution.jar VALIDATION_STRINGENCY= SILENT I= /dev/stdin O= output_file.txt ASSUME_SORTED= true REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa CHART= output_file.pdf ALIGNED_READS_ONLY= true
4. command name= MeanQualityByCycle,command line= java -Xmx1g -jar /packages/picard-tools/1.89/MeanQualityByCycle.jar VALIDATION_STRINGENCY= SILENT ASSUME_SORTED= true REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa I= /dev/stdin O= output_file.txt CHART= output_file.pdf ALIGNED_READS_ONLY= true
5. command name= CollectGcBiasMetrics,command line= java -Xmx1g -jar /packages/picard-tools/1.89/CollectGcBiasMetrics.jar VALIDATION_STRINGENCY= SILENT I= /dev/stdin O= output_file.txt SUMMARY_OUTPUT= output_file_summary.txt CHART= output_file.pdf ASSUME_SORTED= true REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa
6. command name= CollectRnaSeqMetrics,command line= java -Xmx1g -jar /packages/picard-tools/1.89/CollectRnaSeqMetrics.jar VALIDATION_STRINGENCY= SILENT ASSUME_SORTED= true REF_FLAT= /db/yap/ucsc/may_02_2013/gtf/hg19/human_refflat_for_picard.gff RIBOSOMAL_INTERVALS= /db/yap/ucsc/may_02_2013/gtf/hg19/Homo_sapiens_assembly19.rRNA.interval_list STRAND_SPECIFICITY= NONE I= /dev/stdin O= output_file.txt CHART_OUTPUT= output_file.pdf REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa
7. command name= CalculateHsMetrics,command line= /packages/picard-tools/1.89/CalculateHsMetrics.jar VALIDATION_STRINGENCY= SILENT BAIT_INTERVALS= /accessory_files/TruSeq_exome_targeted_regions_for_picard.bed TARGET_INTERVALS= /accessory_files/TruSeq_exome_targeted_regions_for_picard.bed INPUT= /dev/stdin OUTPUT= output_file.txt REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa
8. command name= CollectTargetedPcrMetrics,command line= java -Xmx1g -jar /packages/picard-tools/1.89/CollectTargetedPcrMetrics.jar VALIDATION_STRINGENCY= SILENT AMPLICON_INTERVALS= /accessory_files/TruSeq_exome_targeted_regions_for_picard.bed TARGET_INTERVALS= /accessory_files/TruSeq_exome_targeted_regions_for_picard.bed INPUT= /dev/stdin OUTPUT= output_file.txt REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa
4. command type= :begin
command input : ['input_file_type *coordinate*', 'input_directory aligner_output']
1. command name= CollectInsertSizeMetrics,command line= java -Xmx1g -jar /packages/picard-tools/1.89/CollectInsertSizeMetrics.jar VALIDATION_STRINGENCY= SILENT ASSUME_SORTED= true I= file_based_input O= output_file.txt H= output_file.pdf TMP_DIR= /scratch/$USER HISTOGRAM_WIDTH= 500
5. command type= :begin
command input : ['input_file_type *coordinate*', 'input_directory aligner_output']
1. command name= MarkDuplicates,command line= java -Xmx1g -jar /packages/picard-tools/1.89/MarkDuplicates.jar VALIDATION_STRINGENCY= SILENT TMP_DIR= /scratch/$USER MAX_RECORDS_IN_RAM= 1000000 MAX_SEQUENCES_FOR_DISK_READ_ENDS_MAP= 2000 ASSUME_SORTED= true I= file_based_input O= output_file.bam METRICS_FILE= output_file.txt
6. command type= :begin
command input : ['input_file_type *coordinate*', 'input_directory aligner_output']
1. command name= cufflinks,command line= /packages/cufflinks/2.1.1/cufflinks file_based_input -o output_directory -p 12 -G /accessory_files/human-ucsc-refGene.gtf
-------------------------------------------------------------------------------------------------------------
******************* YAP CHECK SUMMARY *******************
* --Syntax check : Passed *
* --Compatibility check : Passed *
* --File paths check : Passed With Warnings *
*********************************************************
* YAP Configuration overall check status: Passed With Warnings
--------------------------------------- YAP Check Error/Warning Info ---------------------------------------
-------------------------------------------------------------------------------------------------------------
--YAP Configuration File paths check status: Passed With Warnings
Warning: At Line: 21 in file: bowtie_1.0.0_configuration.cfg. Files were found using basename in /db/nibrgenome/NG00006.0/indexes/bowtie/hg19. Please make sure that command: bowtie can work with basenames.
-------------------------------------------------------------------------------------------------------------
-------------------------- YAP configurations check end for Workflow = yap2.3_test --------------------------
-------------------- PROVENANCE --------------------
PREPROCESS:
/packages/FastQScreen/v0.4.1/fastq_screen --subset 500000 --paired --outdir /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/preprocess_output --conf fastq_screen_v0.4.1.conf --aligner bowtie /db/yap/benchmark/robin_50_samples/RN0000108D_1.fq /db/yap/benchmark/robin_50_samples/RN0000108D_2.fq
/packages/fastqc/0.10.1/fastqc --outdir /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/preprocess_output --extract --threads 12 /db/yap/benchmark/robin_50_samples/RN0000108D_1.fq /db/yap/benchmark/robin_50_samples/RN0000108D_2.fq
ALIGNMENT :
INPUT: /db/yap/benchmark/robin_50_samples/RN0000108D_1.fq and /db/yap/benchmark/robin_50_samples/RN0000108D_2.fq chunk number= 0
/packages/bowtie/1.0.0/bowtie /db/nibrgenome/NG00006.0/indexes/bowtie/hg19 -q -v 2 -k 10 -m 10 --best -S -p 8 -1 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000003008281_0.797636572311_pipe_0_0_1 -2 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000003008281_0.797636572311_pipe_0_0_2 >/test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000000.sam
samtools view -bhS - samtools sort -on -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000000_queryname | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000000_queryname.sam
samtools view -bhS - samtools sort -o -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000000_coordinate | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000000_coordinate.sam
INPUT: /db/yap/benchmark/robin_50_samples/RN0000108D_1.fq and /db/yap/benchmark/robin_50_samples/RN0000108D_2.fq chunk number= 1
/packages/bowtie/1.0.0/bowtie /db/nibrgenome/NG00006.0/indexes/bowtie/hg19 -q -v 2 -k 10 -m 10 --best -S -p 8 -1 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000013008281_0.797636572311_pipe_1_0_1 -2 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000013008281_0.797636572311_pipe_1_0_2 >/test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000001.sam
samtools view -bhS - samtools sort -on -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000001_queryname | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000001_queryname.sam
samtools view -bhS - samtools sort -o -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000001_coordinate | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000001_coordinate.sam
INPUT: /db/yap/benchmark/robin_50_samples/RN0000108D_1.fq and /db/yap/benchmark/robin_50_samples/RN0000108D_2.fq chunk number= 2
/packages/bowtie/1.0.0/bowtie /db/nibrgenome/NG00006.0/indexes/bowtie/hg19 -q -v 2 -k 10 -m 10 --best -S -p 8 -1 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000023008281_0.797636572311_pipe_2_0_1 -2 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000023008281_0.797636572311_pipe_2_0_2 >/test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000002.sam
samtools view -bhS - samtools sort -on -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000002_queryname | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000002_queryname.sam
samtools view -bhS - samtools sort -o -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000002_coordinate | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000002_coordinate.sam
INPUT: /db/yap/benchmark/robin_50_samples/RN0000108D_1.fq and /db/yap/benchmark/robin_50_samples/RN0000108D_2.fq chunk number= 3
/packages/bowtie/1.0.0/bowtie /db/nibrgenome/NG00006.0/indexes/bowtie/hg19 -q -v 2 -k 10 -m 10 --best -S -p 8 -1 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000033008281_0.797636572311_pipe_3_0_1 -2 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000033008281_0.797636572311_pipe_3_0_2 >/test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000003.sam
samtools view -bhS - samtools sort -on -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000003_queryname | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000003_queryname.sam
samtools view -bhS - samtools sort -o -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000003_coordinate | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000003_coordinate.sam
INPUT: /db/yap/benchmark/robin_50_samples/RN0000108D_1.fq and /db/yap/benchmark/robin_50_samples/RN0000108D_2.fq chunk number= 4
/packages/bowtie/1.0.0/bowtie /db/nibrgenome/NG00006.0/indexes/bowtie/hg19 -q -v 2 -k 10 -m 10 --best -S -p 8 -1 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000043008281_0.797636572311_pipe_4_0_1 -2 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000043008281_0.797636572311_pipe_4_0_2 >/test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000004.sam
samtools view -bhS - samtools sort -on -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000004_queryname | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000004_queryname.sam
samtools view -bhS - samtools sort -o -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000004_coordinate | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000004_coordinate.sam
INPUT: /db/yap/benchmark/robin_50_samples/RN0000108D_1.fq and /db/yap/benchmark/robin_50_samples/RN0000108D_2.fq chunk number= 5
/packages/bowtie/1.0.0/bowtie /db/nibrgenome/NG00006.0/indexes/bowtie/hg19 -q -v 2 -k 10 -m 10 --best -S -p 8 -1 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000053008281_0.797636572311_pipe_5_0_1 -2 /scratch/kulkatr1//kulkatr1/yap_temp/aligner_RN0000108D_1_0000053008281_0.797636572311_pipe_5_0_2 >/test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000005.sam
samtools view -bhS - samtools sort -on -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000005_queryname | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000005_queryname.sam
samtools view -bhS - samtools sort -o -m 100000000 - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000005_coordinate | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000005_coordinate.sam
MERGE ALIGNMENT OUTPUT :
samtools merge -n - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000002_queryname.bam /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000003_queryname.bam /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000005_queryname.bam /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000000_queryname.bam /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000001_queryname.bam /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000004_queryname.bam | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_queryname.sam
samtools merge - /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000005_coordinate.bam /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000001_coordinate.bam /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000000_coordinate.bam /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000002_coordinate.bam /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000003_coordinate.bam /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/aligner_RN0000108D_1_000004_coordinate.bam | samtools view -h - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_coordinate.sam
POSTPROCESS :
INPUT: /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_queryname.sam
/packages/python/2.6.5_gnu/bin/htseq-count -s no -q /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_queryname.sam /accessory_files/human-ucsc-refGene.gtf >/test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_htseq-count.out
INPUT: /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_coordinate.sam
yap_exon_count -f 1.0 -exon_coordinates_file /accessory_files/human-ucsc-final_exon_coord.bed -exon_CoordToNumber_file /accessory_files/human-ucsc-final_exon_coord_number.bed -i - -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_yap_exon_count
java -Xmx1g -jar /packages/picard-tools/1.89/CollectAlignmentSummaryMetrics.jar VALIDATION_STRINGENCY= SILENT I= /dev/stdin O= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_CollectAlignmentSummaryMetrics.txt IS_BISULFITE_SEQUENCED= true ASSUME_SORTED= True REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa QUIET= True
java -Xmx1g -jar /packages/picard-tools/1.89/QualityScoreDistribution.jar VALIDATION_STRINGENCY= SILENT I= /dev/stdin O= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_QualityScoreDistribution.txt ASSUME_SORTED= true REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa CHART= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_QualityScoreDistribution.pdf ALIGNED_READS_ONLY= true
java -Xmx1g -jar /packages/picard-tools/1.89/MeanQualityByCycle.jar VALIDATION_STRINGENCY= SILENT ASSUME_SORTED= true REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa I= /dev/stdin O= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_MeanQualityByCycle.txt CHART= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_MeanQualityByCycle.pdf ALIGNED_READS_ONLY= true
java -Xmx1g -jar /packages/picard-tools/1.89/CollectGcBiasMetrics.jar VALIDATION_STRINGENCY= SILENT I= /dev/stdin O= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_CollectGcBiasMetrics.txt SUMMARY_OUTPUT= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_CollectGcBiasMetrics_summary.txt CHART= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_CollectGcBiasMetrics.pdf ASSUME_SORTED= true REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa
java -Xmx1g -jar /packages/picard-tools/1.89/CollectRnaSeqMetrics.jar VALIDATION_STRINGENCY= SILENT ASSUME_SORTED= true REF_FLAT= /db/yap/ucsc/may_02_2013/gtf/hg19/human_refflat_for_picard.gff RIBOSOMAL_INTERVALS= /db/yap/ucsc/may_02_2013/gtf/hg19/Homo_sapiens_assembly19.rRNA.interval_list STRAND_SPECIFICITY= NONE I= /dev/stdin O= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_CollectRnaSeqMetrics.txt CHART_OUTPUT= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_CollectRnaSeqMetrics.pdf REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa
/packages/picard-tools/1.89/CalculateHsMetrics.jar VALIDATION_STRINGENCY= SILENT BAIT_INTERVALS= /accessory_files/TruSeq_exome_targeted_regions_for_picard.bed TARGET_INTERVALS= /accessory_files/TruSeq_exome_targeted_regions_for_picard.bed INPUT= /dev/stdin OUTPUT= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_CalculateHsMetrics.txt REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa
java -Xmx1g -jar /packages/picard-tools/1.89/CollectTargetedPcrMetrics.jar VALIDATION_STRINGENCY= SILENT AMPLICON_INTERVALS= /accessory_files/TruSeq_exome_targeted_regions_for_picard.bed TARGET_INTERVALS= /accessory_files/TruSeq_exome_targeted_regions_for_picard.bed INPUT= /dev/stdin OUTPUT= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_CollectTargetedPcrMetrics.txt REFERENCE_SEQUENCE= /db/nibrgenome/NG00006.0/fasta/hg19.fa
INPUT: /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_coordinate.sam
java -Xmx1g -jar /packages/picard-tools/1.89/CollectInsertSizeMetrics.jar VALIDATION_STRINGENCY= SILENT ASSUME_SORTED= true I= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_coordinate.sam O= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_CollectInsertSizeMetrics.txt H= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_CollectInsertSizeMetrics.pdf TMP_DIR= /scratch/$USER HISTOGRAM_WIDTH= 500
INPUT: /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_coordinate.sam
java -Xmx1g -jar /packages/picard-tools/1.89/MarkDuplicates.jar VALIDATION_STRINGENCY= SILENT TMP_DIR= /scratch/$USER MAX_RECORDS_IN_RAM= 1000000 MAX_SEQUENCES_FOR_DISK_READ_ENDS_MAP= 2000 ASSUME_SORTED= true I= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_coordinate.sam O= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_MarkDuplicates.bam METRICS_FILE= /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output/RN0000108D_1_MarkDuplicates.txt
INPUT: /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_coordinate.sam
/packages/cufflinks/2.1.1/cufflinks /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/aligner_output/RN0000108D_1_coordinate.sam -o /test_output/yap2.3_test/RN0000108D_1/no_barcode_specified/postprocess_output -p 12 -G /accessory_files/human-ucsc-refGene.gtf
--------------------Analysis End Time For Workflow : yap2.3_test 2014/09/04 19:43:14-------------------- Requirements
YAP only runs on Linux systems!
Dependencies:
The following dependencies have to be first installed in your environment. Once installed, make sure these dependencies are added to your path.
- Recent versions of gcc (gcc 4.8.x is well tested)
- Python 2.7.7
- Openmpi 1.6.5
- Python modules:
- MPI4py - 1.3
- PyPdf - 1.13
- Numpy - 1.7.1
- netsa-utils - 1.4.3
- bedtools - 2.15.0
- samtools - 0.1.18
System Configuration:
YAP provides a framework to run external tools and data, so the tools used in the workflows drive the system requirements. It can be installed on multicore linux workstation with a decent amount of memory for small data, or on large cluster systems to scale optimally for large data processing. The framework has been tested extensively for NGS data on clusters with minimum system configuration of 8-12 cores and 24-48 GB memory.
YAP Setup
- Download the yap source from here
-
Uncompress the source directory
for example: uncompress the directory as
/home/packages/YAP -
Set
YAP_HOMEenvironment variable to the source directory.$ export YAP_HOME=/home/packages/YAP -
Add bin directory to path
$ export PATH=$PATH:$YAP_HOME/bin/ -
Set
YAP_LOCAL_TEMPDIRenvironment variable for temporary computation. For optimum performance point this directory to a location which is local to the machine.$ export YAP_LOCAL_TEMPDIR=/scratch/username/yap_temp
Verification
$ echo $YAP_HOME
/home/packages/YAP
$ echo $YAP_LOCAL_TEMPDIR
/scratch/username/yap_temp
$ which yap
/home/packages/YAP/bin/yap
Running a YAP job
Once you've set your environment, it is best to run a quick demo job to get the feel of running YAP. The following section is meant to be interactive and hence you would need Linux account access and access to the cluster.
After downloading the project, please see the demo configuration files in yap/cfg.
There are 3 stages in YAP - Preprocess, Alignment and Postprocess. You can have command level control of these three stages in the namesake configuration files and a workflow level control in the workflow_configuration.
| Configuration | Purpose |
|---|---|
| aligner_configuration | bwa, bowtie, bowtie2, tophat or insert your own aligner |
| postprocess_configuration | postalignment packages, generate counts or metrics |
| preprocess_configuration | pre-alignment packages to massage your seqdata |
| workflow_configuration | manage metadata, specify input files, paths and output directories |
| yap_sge | submitting your job to the cluster |
The demo runs a RNASeq QC and counts workflow consisting of:
- Preprocess: FastQC, Fastqscreen
- Alignment: Bowtie, both queryname and coordinate sorted
- Postprocess: yap junction and exon counts, Picard tools (PostQC), HTSeq (Raw counts) and Cufflinks (normalized counts)
We run this workflow on 2 nodes on the UGE cluster.
To run the yap_demo job, we next need to check to see if our configuration files are correct using the command.
cd <your_working directory>
yap --check workflow_configuration.cfg
The yap --check command checks to see
- If all paths specified are valid
- If YAP finds the appropriate input files
- Checks for syntax errors
- Lists commands to be executed.
- Gives section-wise error/warning report.
Running the YAP job
mpirun -n <number_of_cores> yap workflow_configuration.cfg
If you have a SGE environment, pass the number of slots into the $NSLOTS variable.
Interested in Novartis Engineering?
At Novartis, you'll be at the forefront of technology — helping to shape it, develop it, and make it impactful. Partnering with scientists, our engineers create cutting-edge, state-of-the-art solutions that accelerate drug discovery and ultimately improve patients' lives.
Learn more